CrystalFlow: Revolutionizing Crystal Structure Prediction with Flow-Based Generative Models
In the rapidly advancing field of materials science, the accurate prediction of crystal structures remains one of the grand challenges. The arrangement of atoms within a crystal directly determines its electronic, mechanical, and chemical properties, and mastering the art of predicting these arrangements could unlock a new era of materials discovery — from superconductors to battery electrodes and quantum materials.
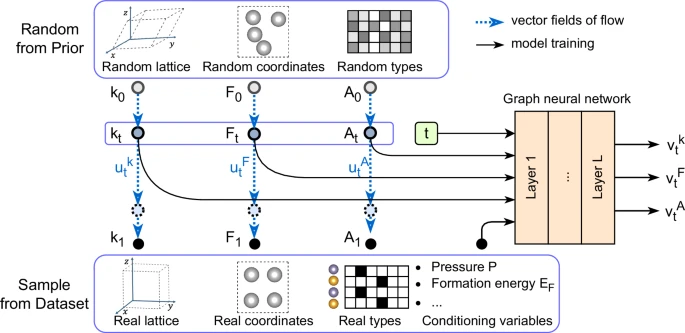
A newly published study in Nature Communications by Xiaoshan Luo and colleagues from Jilin University and the Chinese Academy of Sciences introduces CrystalFlow, a groundbreaking flow-based generative model that redefines the computational efficiency and accuracy of crystal structure prediction (CSP). This innovative approach leverages Continuous Normalizing Flows (CNFs) and Conditional Flow Matching (CFM) — two emerging deep learning frameworks that allow smooth and invertible transformations of probability distributions — to generate realistic crystal structures with unprecedented precision.
The Challenge of Crystal Structure Prediction
CSP traditionally involves searching for the most stable arrangement of atoms within a potential energy landscape. This process is computationally intensive because the number of possible configurations grows exponentially with system size. Methods like USPEX, CALYPSO, and particle swarm optimization have pushed the field forward, but even with quantum-mechanical approaches such as density functional theory (DFT), predicting new stable materials remains extremely challenging.
Recent years have seen the rise of AI-driven generative models for materials discovery, using architectures inspired by image and text generation — including Variational Autoencoders (VAE), Generative Adversarial Networks (GANs), and Diffusion Models. However, diffusion models, while powerful, are often limited by their need for large integration steps and high computational cost.
What Makes CrystalFlow Different
CrystalFlow stands out by introducing a flow-based modeling framework that is not only more computationally efficient — requiring ten times fewer integration steps than diffusion-based approaches — but also symmetry-aware. It incorporates the intrinsic periodic, rotational, and permutation symmetries of crystalline systems through equivariant graph neural networks (GNNs). This ensures that the generated structures respect the fundamental physics of materials.
In benchmark tests on the MP-20 and MPTS-52 datasets, which include tens of thousands of experimentally known crystal structures, CrystalFlow achieved accuracy metrics comparable or superior to state-of-the-art diffusion models such as DiffCSP and FlowMM. On the larger and more complex MP-CALYPSO-60 dataset, CrystalFlow demonstrated excellent scalability and was able to generate pressure-dependent crystal structures, crucial for studying materials under extreme conditions.
Generative AI for Materials Design
Beyond structure prediction, CrystalFlow also excels in de novo materials generation — the creation of new, previously unseen compounds. When trained to condition on specific properties such as formation energy or pressure, the model can generate entirely new crystalline arrangements optimized for those parameters. After DFT relaxation, more than 90% of generated structures were new and physically stable, demonstrating the model’s potential as an intelligent engine for inverse materials design.
The authors highlight that the flow-based generative architecture used by CrystalFlow enables efficient sampling from high-dimensional distributions. In other words, it can "learn" the statistical physics of how atoms prefer to arrange themselves — something that traditional optimization algorithms struggle to achieve without enormous computational effort.
A Glimpse into the Future of AI-Powered Materials Discovery
The implications of CrystalFlow extend far beyond theoretical modeling. As computational tools become increasingly integrated with experimental materials design, AI models like CrystalFlow could drastically accelerate the development of superconductors, thermoelectric materials, hydrogen-storage compounds, and next-generation semiconductors. By reducing computational bottlenecks and increasing predictive accuracy, flow-based frameworks may soon become indispensable in both academic research and industrial applications.
Looking ahead, the authors envision combining CrystalFlow with transformer-based architectures or large language models (LLMs) to further enhance property prediction and multi-modal learning capabilities. Such hybrid AI systems could one day autonomously design, test, and optimize entirely new classes of materials.
Conclusion
CrystalFlow represents a new milestone in AI-driven materials science — an elegant synthesis of mathematical rigor, physical insight, and deep learning innovation. By merging flow-based generative modeling with crystallographic symmetry principles, the research sets a powerful precedent for the future of computational materials discovery.
You can read the full original article on Nature Communications here: https://www.nature.com/articles/s41467-025-64364-4
This article was prepared for Quantum Server Networks with the help of advanced AI technologies to enhance clarity, structure, and SEO optimization.
Sponsored by PWmat (Lonxun Quantum) – a leading developer of GPU-accelerated materials simulation software for cutting-edge quantum, energy, and semiconductor research. Learn more at: https://www.pwmat.com/en
📘 Download our latest brochure: PWmat PDF Brochure
🎁 Try PWmat for free! Request a trial version tailored to your research needs: Request a Free Trial
📞 Phone: +86 400-618-6006
📧 Email: support@pwmat.com
🌐 Connect with us on Social Media:
#AIinMaterialsScience #CrystalFlow #GenerativeAI #MaterialsDiscovery #QuantumServerNetworks #MachineLearning #Crystallography #NatureCommunications #FlowModel #CSP #DeepLearningMaterials #PWmat #AIResearch #MaterialsSimulation


Comments
Post a Comment