Capturing Short-Range Order in High-Entropy Alloys with Machine Learning Potentials
High-entropy alloys (HEAs) are revolutionizing the field of materials science by combining multiple metallic elements in nearly equal proportions. This seemingly random distribution of atoms leads to extraordinary mechanical properties such as high fracture resistance, exceptional toughness, and resistance to deformation. However, the microscopic structural features that govern these properties are not entirely random — they are influenced by chemical short-range order (SRO), a tendency for certain atomic arrangements to occur more frequently than others.
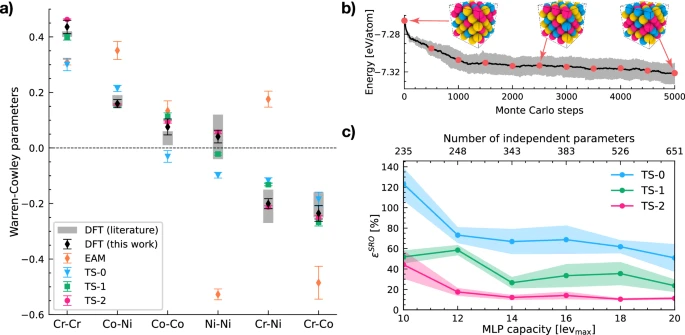
Machine learning potentials provide new insights into chemical short-range order in CrCoNi high-entropy alloys.
A recent study by Yifan Cao, Killian Sheriff, and Rodrigo Freitas, published in npj Computational Materials, introduces a systematic approach to capturing SRO in high-entropy alloys using machine learning interatomic potentials (MLPs). Their research focuses on the CrCoNi alloy, a benchmark system in the study of HEAs, which has already demonstrated remarkable damage tolerance even at cryogenic temperatures.
Why Short-Range Order Matters
Chemical short-range order influences a range of fundamental properties in metallic alloys. It impacts:
- Dislocation mobility, which governs how metals deform under stress.
- Grain boundary stability, affecting strength and durability.
- Stacking-fault energy, a crucial parameter for predicting deformation mechanisms such as twinning and slip.
- Phase stability, determining whether a material favors face-centered cubic (fcc) or hexagonal close-packed (hcp) crystal structures.
Traditional computational methods such as density-functional theory (DFT) struggle to capture SRO at realistic length scales due to computational limits. This is where machine learning potentials become powerful — they enable simulations with thousands of atoms while preserving the quantum-level accuracy of DFT.
The Machine Learning Approach
The authors employ Moment Tensor Potentials (MTPs), a class of MLPs known for their ability to systematically improve in accuracy. By carefully designing training datasets that capture the complexity of atomic motifs in CrCoNi, the researchers develop potentials that not only predict energies and forces but also reproduce subtle short-range ordering effects.
Their systematic training strategy progresses from random chemical configurations to more physically informed datasets derived from DFT Monte Carlo simulations. This leads to models (such as their TS-2 and TS-3 datasets) capable of stabilizing both the crystal and liquid phases of CrCoNi — a major breakthrough in simulating these complex systems.
Key Findings
The study demonstrates that:
- Energy accuracy alone is insufficient — models must be tested on their ability to reproduce material properties such as stacking-fault energy and phase stability.
- Machine learning potentials trained with targeted chemical motifs significantly outperform those trained on random data alone.
- Large-scale simulations (up to ~10,000 atoms) are required to fully capture the length scales of SRO, beyond the reach of traditional DFT.
- SRO strongly influences temperature-dependent stacking-fault energies and fcc–hcp phase stability, providing new insights into mechanical behavior.
Broader Implications
The ability to capture short-range order using machine learning opens the door to rational materials design. By better understanding the interplay between atomic structure and mechanical properties, scientists can engineer high-performance alloys for aerospace, energy, and quantum technologies. Moreover, the design principles outlined in this work are generalizable to a wide variety of metallic systems, including ordered compounds and defect-rich materials.
Conclusion
This research highlights a new paradigm: accurate modeling of high-entropy alloys requires more than just raw computational accuracy — it requires physically meaningful training data that reflects the chemical complexity of real materials. Machine learning potentials, when designed with this principle in mind, provide unprecedented fidelity in simulating the properties of HEAs.
Footnote: This blog article was prepared with the assistance of AI technologies.
Sponsored by PWmat (Lonxun Quantum) – a leading developer of GPU-accelerated materials simulation software for cutting-edge quantum, energy, and semiconductor research. Learn more about our solutions at: https://www.pwmat.com/en
📘 Download our latest company brochure to explore our software features, capabilities, and success stories: PWmat PDF Brochure
🎁 Interested in trying our software? Fill out our quick online form to request a free trial and receive additional information tailored to your R&D needs: Request a Free Trial and Info
📞 Phone: +86 400-618-6006
📧 Email: support@pwmat.com
Original research source: npj Computational Materials
Hashtags: #MaterialsScience #HighEntropyAlloys #MachineLearning #QuantumMaterials #ComputationalMaterials #AlloyDesign #Nanotechnology #PWmat


Comments
Post a Comment