Breaking Barriers in Surface Chemistry: The autoSKZCAM Framework for Ionic Materials
Published on Quantum Server Networks
Understanding and predicting chemical reactions on surfaces lies at the heart of modern materials science. From heterogeneous catalysis to energy storage and greenhouse gas sequestration, surface chemistry defines the efficiency and viability of advanced technologies. Yet, computationally modeling these processes with both accuracy and efficiency has been a grand challenge.
A recent study published in Nature Chemistry introduces a breakthrough: the autoSKZCAM framework, an automated and open-source method that applies correlated wavefunction theory (cWFT) to surfaces of ionic materials at costs comparable to density functional theory (DFT). This achievement not only bridges the accuracy gap but also enables routine, large-scale studies of surface processes with chemical accuracy.
The Challenge: Accuracy vs. Efficiency in Surface Modeling
For decades, density functional theory has been the workhorse of computational surface science. Its ability to capture essential physics at moderate computational cost has enabled countless insights into adsorption, catalysis, and energy storage. However, DFT’s approximations often introduce inconsistencies and fail to provide systematically improvable accuracy. Different DFT functionals can even yield contradictory results for the same system, complicating the search for reliable predictions.
By contrast, correlated wavefunction methods—such as coupled-cluster theory CCSD(T)—are considered the “gold standard” in quantum chemistry. They deliver benchmark-level accuracy but at costs that have historically made them impractical for realistic surface problems involving large clusters and complex adsorbates. Until now, only a handful of simple adsorbate–surface systems could be treated at this level, leaving a significant gap in predictive modeling capabilities.
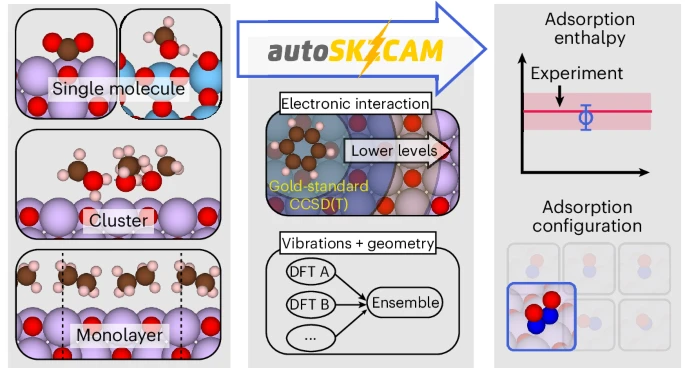
Introducing autoSKZCAM: Automation Meets Accuracy
The autoSKZCAM framework, developed by an international collaboration led by researchers at the University of Cambridge, TU Wien, Princeton, and others, solves this long-standing bottleneck. The framework employs a divide-and-conquer embedding approach: partitioning the adsorption enthalpy (Hads) into contributions that are treated at different levels of theory. The most critical interaction energy terms are calculated with local CCSD(T), while more routine vibrational and thermal contributions are handled with ensembles of DFT functionals.
Crucially, the entire workflow is automated and open source, enabling researchers to obtain high-quality predictions without requiring deep expertise in cWFT implementations. The method has been coded into a package available on GitHub, making it a powerful tool for both academia and industry.
Validating Against Experiment
In their study, the authors applied autoSKZCAM to 19 adsorbate–surface systems, spanning molecules such as CO, NO, N2O, NH3, H2O, CO2, CH3OH, and benzene on surfaces like MgO(001) and TiO2. The framework consistently reproduced experimental adsorption enthalpies within error bars, achieving a level of agreement unmatched by previous theoretical approaches.
This capability also helped resolve longstanding debates. For instance, the correct adsorption geometry of NO on MgO(001)—disputed across many DFT studies—was clarified by autoSKZCAM, which identified the stable dimer configuration in agreement with spectroscopic evidence. Similarly, the framework confirmed that CO2 binds in a chemisorbed carbonate state on MgO, settling a controversy between physisorption and chemisorption interpretations.
A Benchmark for the Future
Beyond reproducing known experiments, the framework has created a benchmark dataset, dubbed Surf13, offering CCSD(T)-quality reference values for adsorption on ionic surfaces. This dataset is invaluable for testing and improving new DFT functionals, as well as training emerging machine-learning approaches to materials modeling. By providing reliable baselines, autoSKZCAM accelerates the development of computational tools across surface science and catalysis research.
Implications for Catalysis, Energy, and Climate Solutions
The implications are vast. Catalysts for clean fuel production, adsorbents for CO2 capture, and electrodes for next-generation batteries all rely on accurate knowledge of adsorption energies and configurations. With autoSKZCAM, researchers can now explore larger systems and more complex adsorption scenarios with confidence, cutting down costly trial-and-error cycles in experimental design.
Moreover, because the framework’s costs approach those of hybrid DFT, it is feasible to incorporate into high-throughput computational pipelines, enabling systematic screening of candidate materials for catalysis and energy storage. This positions autoSKZCAM as a cornerstone of future computational materials discovery.
Looking Ahead
While the current framework is optimized for ionic materials such as metal oxides, ongoing work aims to extend its reach to covalent and metallic surfaces. Integration with machine-learned potentials could further broaden its applicability to finite temperature dynamics and large-scale simulations.
Ultimately, autoSKZCAM represents a paradigm shift—transforming correlated quantum chemistry from a specialist’s tool into a broadly accessible, automated resource for the entire materials science community.
Original research article: An accurate and efficient framework for modelling the surface chemistry of ionic materials, Nature Chemistry (2025).
Footnote: This blog post was prepared with the assistance of AI technologies for content generation and optimization.
Sponsored by PWmat (Lonxun Quantum) – a leading developer of GPU-accelerated materials simulation software for cutting-edge quantum, energy, and semiconductor research. Learn more about our solutions at: https://www.pwmat.com/en
📘 Download our latest company brochure to explore our software features, capabilities, and success stories: PWmat PDF Brochure
🎁 Interested in trying our software? Fill out our quick online form to request a free trial and receive additional information tailored to your R&D needs: Request a Free Trial and Info
📞 Phone: +86 400-618-6006
📧 Email: support@pwmat.com
#SurfaceChemistry #ComputationalMaterialsScience #QuantumChemistry #Catalysis #EnergyStorage #CO2Capture #DFT #CCSDT #MachineLearningMaterials


Comments
Post a Comment