Revolutionizing Molecular Property Prediction in Data-Scarce Regimes with Adaptive AI
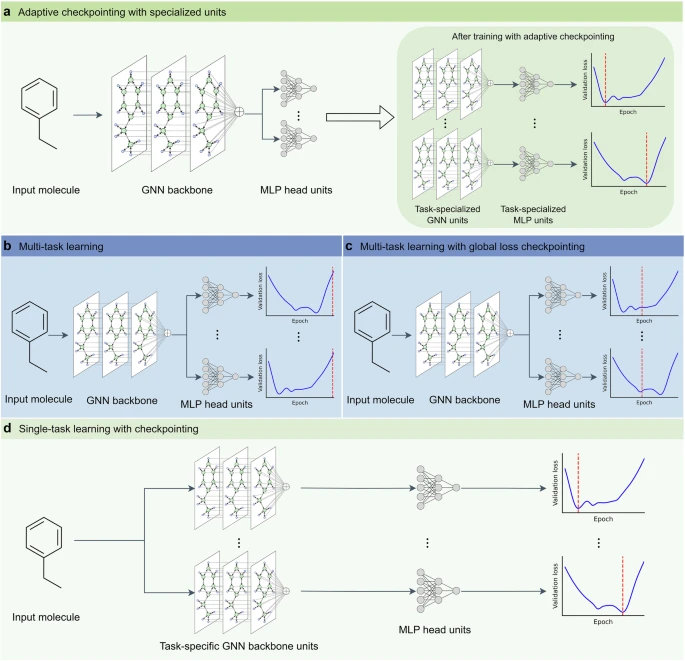
In the rapidly advancing field of materials science, predicting molecular properties with high accuracy is essential for breakthroughs in pharmaceuticals, green energy, polymers, and sustainable fuels. However, data scarcity often limits the effectiveness of traditional machine learning models, which require vast labeled datasets to perform well. A recent study published in Communications Chemistry introduces an innovative approach called Adaptive Checkpointing with Specialization (ACS), designed to address this challenge and accelerate AI-driven molecular design even in ultra-low-data environments.
Read the original article here
Overcoming Data Scarcity in Molecular Prediction
Machine learning-based molecular property prediction models have transformed our ability to explore chemical space and design high-performance materials. Yet, their predictive power often depends on the availability of large, high-quality datasets. This limitation is particularly severe in fields like sustainable aviation fuels (SAFs), where experimental data collection is time-consuming and costly. Multi-task learning (MTL), which leverages correlations among related properties, has been proposed to ease these bottlenecks. But MTL faces its own challenge: negative transfer (NT)—where training on multiple tasks can harm rather than help performance.
The ACS framework addresses this issue by combining a shared graph neural network (GNN) backbone with task-specific learning heads. This innovative architecture allows the model to balance shared learning across tasks while isolating individual tasks from detrimental updates caused by NT.
What Makes ACS a Game Changer?
In their study, researchers from King Abdullah University of Science and Technology demonstrated ACS’s ability to achieve accurate predictions with as few as 29 labeled samples. This is an unprecedented advancement for AI in molecular sciences. When applied to predicting 15 key properties of SAF molecules, ACS outperformed existing methods, showing an average improvement of 12.9% and a 32.7% reduction in performance variability.
Key features of ACS include:
- Adaptive Checkpointing: Saves model parameters when individual tasks reach new performance milestones, mitigating negative transfer.
- Task-Specific Heads: Allows specialized learning for each molecular property while still benefiting from shared representations.
- Ultra-Low-Data Learning: Enables robust prediction even in highly imbalanced datasets, typical in real-world scientific applications.
Broader Implications for Materials Discovery
This approach is not limited to SAFs. ACS has significant potential across various domains of materials science and drug discovery where data scarcity and imbalanced datasets are common. By lowering the barrier for accurate AI predictions, ACS paves the way for accelerated development cycles, reduced experimental costs, and the discovery of novel materials with unprecedented properties.
As data-driven innovation becomes a cornerstone of modern science, frameworks like ACS are critical for ensuring that even researchers with limited datasets can harness the power of machine learning.
Future Directions
Future research will likely focus on refining ACS to handle even more complex datasets and integrating it with meta-learning or pre-trained models. These developments could further enhance its ability to generalize across chemical spaces and make it applicable to large-scale industrial challenges.
This breakthrough underscores a transformative era in computational chemistry, where adaptive AI systems can make high-accuracy predictions even with minimal data—a true leap forward for materials innovation.
To explore this study in detail, visit the original article: Molecular property prediction in the ultra-low data regime.
Sponsored by PWmat (Lonxun Quantum) – a leading developer of GPU-accelerated materials simulation software for cutting-edge quantum, energy, and semiconductor research. Learn more about our solutions at: https://www.pwmat.com/en
📘 Download our latest company brochure to explore our software features, capabilities, and success stories: PWmat PDF Brochure
📞 Phone: +86 400-618-6006
📧 Email: support@pwmat.com
#MolecularPrediction #MaterialsScience #MachineLearning #ArtificialIntelligence #GraphNeuralNetworks #SustainableFuels #QuantumServerNetworks



Comments
Post a Comment