Machine Learning Meets Mechanisms: Revolutionizing Molecular Site-Selectivity Prediction
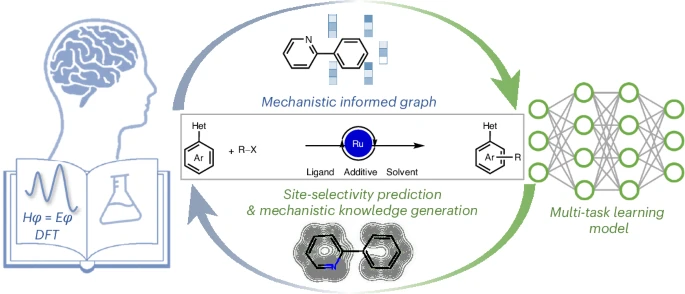
Imagine if predicting the precise site of a chemical reaction on an aromatic ring could be done with machine precision — and with built-in chemical reasoning. Thanks to a groundbreaking new study published in Nature Synthesis, that dream is now closer to reality. A team of chemists and computer scientists has introduced a powerful multitask graph neural network (MT-GNN) that integrates machine learning with density functional theory (DFT) to predict site-selectivity in ruthenium-catalyzed C–H functionalization reactions.
The Challenge: Where Will the Reaction Happen?
Controlling the exact location — or site — where a reaction occurs on an arene is vital in designing molecules for pharmaceuticals, agrochemicals, and materials. Traditionally, this has relied on deep mechanistic understanding, theoretical models, and a fair amount of trial-and-error. But these approaches are limited by human interpretation and the constraints of smaller datasets.
The Breakthrough: A Multitask GNN with Mechanistic Insight
The authors designed a multitask learning framework combining physical property prediction with site-selectivity classification, all powered by a graph neural network. Crucially, their model incorporates mechanism-informed molecular graphs, embedding descriptors like atomic charges and Fukui indices directly into the learning pipeline.
The result? The MT-GNN achieved a remarkable accuracy of 93.4% on a diverse dataset of 256 reactions. It even extrapolated well to new reaction conditions and unfamiliar aromatic scaffolds such as indoles and fused ring systems.
Interpretability Built In: What the Model "Thinks"
Far from being a black box, the MT-GNN also offers interpretability. By visualizing attention weights, the authors identified how different atoms in the arenes contribute to site-selectivity. In some cases, the neural network even suggested new mechanistic hypotheses, which were validated using DFT.
Implications for the Future
This research doesn’t just improve predictions — it represents a shift in how we do chemistry. By uniting AI and quantum chemistry, this work exemplifies how machine learning models can become not only predictive tools, but engines for chemical discovery and theory generation.
For synthetic chemists, computational scientists, and anyone exploring the frontier of reaction modeling, this work is a clear signal: the future of chemical design is intelligent, interpretable, and data-driven.
Dive deeper into the full article on Nature Synthesis.
Source: Chen, Zhang, Hong & Ackermann. Nature Synthesis (2025)
Posted by: Quantum Server Networks


Comments
Post a Comment